We’re starting the series on the 11 principles of ICH E6(R3).
E6(R3) consolidates and reframes the 13 principles from E6(R2) into 11 with clearer emphasis and more practical expectations.
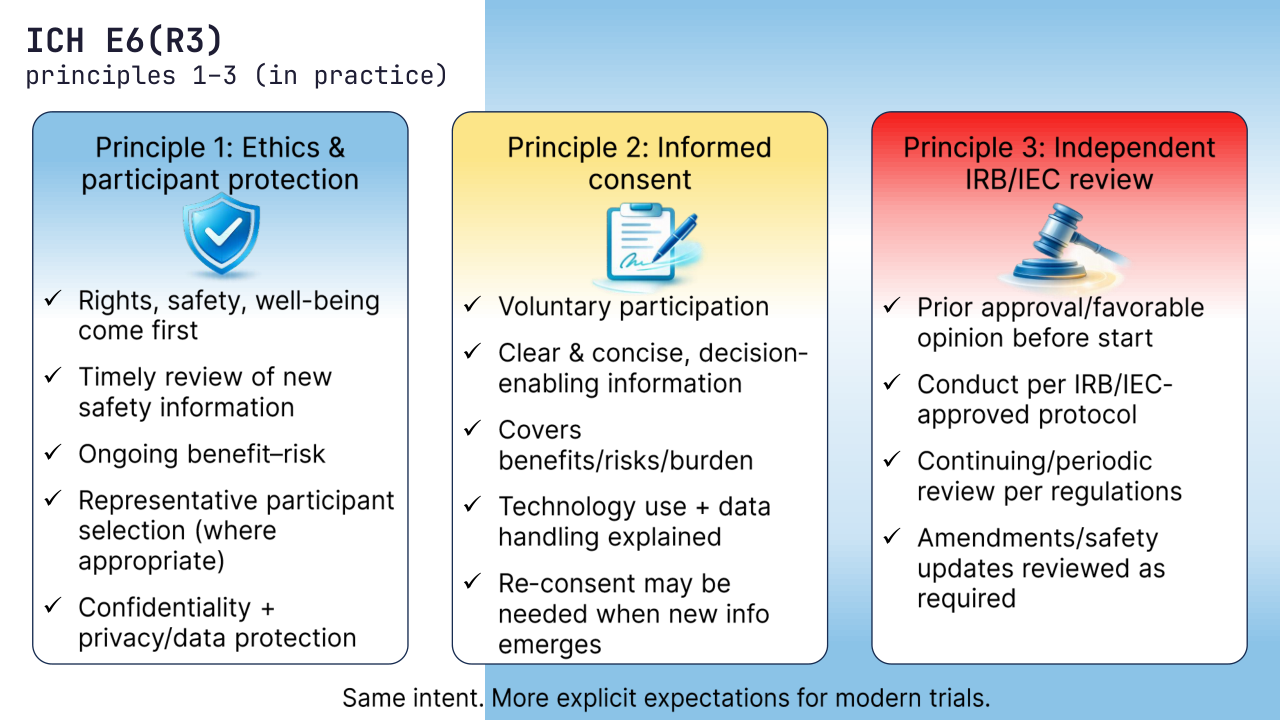
1) Ethical conduct and participant protection (Declaration of Helsinki)
Clinical trials must follow ethical principles, applicable regulations, and always prioritize participants’ rights, safety, and well-being.
R3 makes it more explicit that ethics is continuous: timely review of new safety information, an ongoing benefit–risk view, representative participant selection (where appropriate), and protection of confidentiality/privacy.
🧩Data and stats angle: privacy-by-design and “collect only what you can justify” are part of ethics – not an IT afterthought.
2) Informed consent
Participation must be voluntary and based on a consent process that is clear, concise, and decision-enabling.
R3 pushes beyond formality: consent should reflect trial reality – benefits/risks/burden, technology use (eConsent/ePRO/DHT), data handling, and what happens if a participant withdraws. When new information could affect willingness to continue, re-consent may be needed, not just a document update.
3) Independent review (IRB/IEC)
Trials must be reviewed by an independent IRB/IEC.
Key focus in R3: prior approval/favorable opinion before start, conduct in compliance with the IRB/IEC-approved protocol, and continuing/periodic review per regulations (including review of amendments and key safety updates as required).
🧩Biostat hook: changes that affect endpoints, risks, or data collection often trigger ethics review and can cascade into SAP updates and re-validation of analysis pipelines.
Next: Principles 4-6 – scientific soundness, qualified teams, and quality by design.
Question: what’s hardest in real projects today – representativeness, consent clarity (incl. tech), or IRB/IEC timelines?